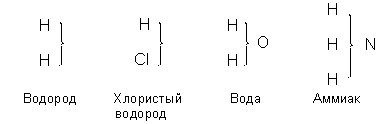|
Краткий очерк
истории химии:
|
Учебное пособие для студентов химфака РГУ
Возникновение структурной химии – Структурные теории – Стереохимия – Координационная химия Возникновение структурной химии В первой половине XIX века зародилась принципиально новая концепция химии – структурная химия, исходящая из предпосылки о том, что свойства вещества определяются не только его составом, но и структурой, т.е. порядком соединения атомов и их пространственным расположением. Самые первые структурные представления с необходимостью возникают вместе с атомистикой Дальтона. Развивая представления о способах объединения "простых атомов" в "сложные атомы", Дальтон преследовал лишь одну цель – создать теорию для объяснения эмпирически открытых стехиометрических законов. Тем не менее, сами избранные Дальтоном символы химических элементов предполагали при изображении сложных атомов выбор определённого порядка соединения атомов между собой. Однако вопрос о порядке соединения атомов оказался отложен на довольно долгое время, поскольку химики не имели никаких фактов, указывающих на влияние способа соединения атомов на свойства вещества. Химическая символика Берцелиуса позволила обойти этот вопрос, хотя и в электрохимической теории Берцелиуса всё же рассматриваются некоторые проблемы ("силы сцепления", "соположение" и т.п.), ставшие впоследствии фундаментальными вопросами структурной химии. Возникновение структурной химии следует, видимо, связывать с открытием явления изомерии. В 1825 г. Иоганн Юстус фон Либих обнаружил, что элементный состав гремучей (фульминовой) кислоты в точности соответствует составу циановой кислоты, которую за год до этого получил Фридрих Вёлер. Повторные анализы, проведённые Вёлером и Либихом, однозначно установили существование веществ, одинаковых по составу, но различающихся по свойствам. Продолжая работы с циановой кислотой, Вёлер, выпаривая раствор изоцианата аммония, получил в 1828 г. изомерное органическое вещество – мочевину. В 1830 г. Й. Я. Берцелиус установил, что виноградная и виннокаменная кислоты также имеют одинаковый состав, но различаются по свойствам. Берцелиус предложил для обнаруженного явления термин "изомерия" (от греческого ισοζ μερον – равной меры). Вскоре обнаружилось, что это явление чрезвычайно распространено в органической химии. В состав органических веществ входит относительно небольшое число элементов – углерод, водород, азот, кислород, сера и фосфор (т.н. элементы-органогены) – при огромном разнообразии свойств. Именно поэтому на протяжении почти всего XIX века структурные представления оказались востребованы, прежде всего, в органической химии. Следует подчеркнуть, однако, что категорически не следует отождествлять понятия "структурная химия" и "органическая химия". В основу решения вопроса о строении органических веществ было положено представления Берцелиуса о радикалах – полярных группах атомов (не содержащих кислорода), способных переходить из одних веществ в другие без изменения. Еще в 1810-1811 гг. Жозеф Луи Гей-Люссак и Луи Жак Тенар показали, что цианидный радикал CN ведёт себя как единичный атом (причём весьма сходный с атомом хлора или брома). Представления о радикалах, хорошо согласующиеся с электрохимической теорией Берцелиуса, позволили распространить эту теорию и на органические вещества. Создание теорий структурной химии Теория сложных радикалов возникла и стала активно разрабатываться многими химиками после работы Либиха и Вёлера "О радикале бензойной кислоты", вышедшей в 1832 г. Либих и Вёлер показали, что группировка атомов С14Н10О2 (правильная брутто-формула – С7Н5О) в цепи превращений бензойной кислоты (бензальдегид – бензойная кислота – бензоилхлорид – бензоилцианид) ведёт себя как единое целое – как некий "органический атом". Теория сложных радикалов быстро получила практически всеобщее признание. В 1837 г. в обобщающей статье "О современном состоянии органической химии", одним из авторов которой являлся Либих, утверждалось, что изучение сложных радикалов – основная задача органической химии, поскольку "циан, амид, бензоил, радикалы аммиака, жиров, алкоголя и его производных образуют истинные элементы органической природы, тогда как простейшие составные части – углерод, водород, кислород и азот – обнаруживаются лишь при разрушении органической материи". Количество описанных радикалов быстро возрастало. Теория сложных радикалов исходила из предположения, что радикалы способны к самостоятельному существованию, хотя химикам и не удавалось их выделить. Берцелиус по этому поводу писал: "Причина, по которой мы не можем изолировать радикалы… не в том, что они не существуют, а в том, что они слишком быстро соединяются". В 1834 г. французский химик Жан Батист Андре Дюма описал явление металепсии – замещения водорода в органических соединениях хлором, при котором сохраняются основные свойства вещества, – получив из этанола (С2Н6О) хлораль (С2Н3Cl3О). Дюма (кстати, соавтор вышеупомянутой статьи Либиха) вначале не придал большого значения открытому явлению, но после получения им в 1839 г. хлоруксусной кислоты пересмотрел свои взгляды. Явление металепсии в корне противоречили электрохимическому дуализму Берцелиуса, попытки которого найти этим экспериментальным фактам какие-либо объяснения в рамках своей теории потерпели неудачу. Металепсия, однако, хорошо согласовывалась с законом изоморфизма Мичерлиха. Дюма в своих работах начал настойчиво развивать мысль о том, что свойства соединений, как показывают экспериментальные факты, определяются только расположением атомов в молекуле, а не их природой. Поэтому, считает Дюма, химикам надлежит заняться выявлением типов расположения атомов в молекуле. "Необходимо сделать вывод о том, что в органической химии существуют типы соединений, которые сохраняются даже в случае замещения в них водорода галогеном". Теория типов Дюма предполагала наличие ряда типов органических соединений, число которых все время увеличивалось: типы спирта, альдегида, кислоты, эфира и т.п. (в принципе типы Дюма можно сопоставить с современным понятием классы органических соединений). Вскоре после создания теории Дюма французские химики Шарль Фредерик Жерар и Огюст Лоран разработали новую теорию типов. В 1840-е годы Жерар и Лоран предложили принципиально новое понимание молекулы химического соединения как унитарной (единой) системы. Высказанное ими предположение о том, что значение электростатических сил преувеличено, вызвало резкую критику со стороны Берцелиуса и других сторонников электрохимического дуализма. Лишь в 1850-х годах (после смерти Берцелиуса) взгляды Жерара и Лорана получили признание. Унитарная теория была изложена Жераром в небольшой книге "Введение в изучение химии по унитарной системе", опубликованной в 1848 г. Суть теории можно свести к следующему: 1. Молекула представляет собой не двойное или тройное тело – совокупность атомов либо радикалов, способных к самостоятельному существованию, но принципиально новую единую систему. 2. Вновь образованное химическое соединение следует рассматривать как полную утрату прежних свойств составившими его элементами. 3. Химическую способность атомов или групп атомов (радикалов) в молекуле можно охарактеризовать с помощью понятия функции, зависящей как от природы атома или группы атомов, так и от природы и количества других атомов (групп атомов). В 1852 г. в работе "Об ангидридах органических кислот" Жерар сформулировал положения, составившие новую теорию типов (теорию типов Жерара-Лорана), которая на основании унитарного подхода включила и представления о сложных радикалах, и идеи Дюма о типах молекул. Согласно новой теории типов, "органические соединения могут быть сведены к трём или четырём типам; каждый из них способен давать <гомологические> ряды…; этими типами являются вода Н2О, водород Н2, хлористый водород HCl и аммиак NН3. Обменивая свой водород на определённые группы, эти типы дают начало кислотам, спиртам, эфирам, гидридам, органическим хлоридам, кетонам, щелочам". Ниже приведены формулы Жерара-Лорана для основных типов:
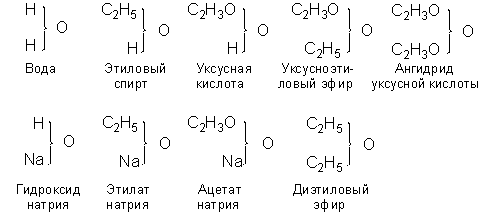
Некоторые соединения, принадлежащие к типу воды и полученные замещением водорода на соответствующие атомы или сложные радикалы, изображались в новой теории типов следующим образом:
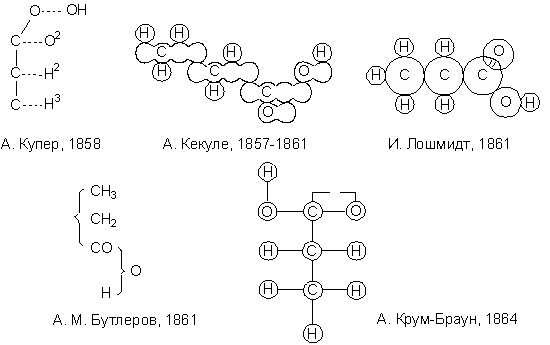
Новая теория типов завоевала в 1850-е годы широкое признание; она позволила систематизировать органические соединения, список которых очень быстро увеличивался и, что особенно важно, привела к открытию многих новых соединений (в частности, существование ангидридов карбоновых кислот было предсказано Жераром на основании данной теории). Новая теория типов развивалась трудами многих учёных: Александр Уильям Уильямсон показал, что простые эфиры можно отнести к типу воды, Шарль Адольф Вюрц исследовал амины, в которых ядром служит атом азота (тип аммиака). В 1854-1855 гг. Фридрих Август Кекуле фон Штрадониц предложил типы сероводорода и (одновременно с Уильямом Одлингом) метана (болотного газа) в качестве побочных типов; Кекуле также начал пользоваться смешанными типами, относя, например, сульфобензойную кислоту к типу воды и водорода. Российский химик Фридрих Бейльштейн опубликовал в 1880 г. обширное руководство по органическим соединениям, размещённым в рациональном порядке с использованием теории Жерара-Лорана. Тем не менее, новая теория типов также не могла полностью удовлетворить химиков. Новая теория типов по-прежнему обходила стороной вопрос о молекулярной структуре. Жерар в рамках унитарного подхода утверждал, что формулы теории типов отражают лишь прошлое и будущее молекулы, но не её строение, которое, по его мнению, можно установить лишь физическими методами. Поэтому Жерар скептически относился к рациональным формулам теории и протестовал против утверждений, что каждому химическому соединению непременно соответствует лишь одна такая формула. Следующим этапом развития структурной химии стала теория валентности, в известной степени представляющая собой отказ от унитарных представлений. Тем не менее, именно теория типов Жерара-Лорана подвела химиков к представлению о единицах сродства атомов и радикалов, на основе которого и была создана теория валентности. Возникновение понятия валентности (атомности) обычно связывают с именем английского химика Эдуарда Франкленда. Франкленд, являвшийся сторонником теории сложных радикалов, изучая металлоорганические соединения, пришёл к выводу о необходимости пересмотреть теорию радикалов и признать некоторые положения теории типов. Развивая высказанные Жераром, Лораном, Одлингом, Уильямсоном и др. идеи о связи между эквивалентностью атомов и эквивалентностью радикалов, Франкленд в 1852 г. предложил ввести понятие "соединительной силы" атомов. Впрочем, хотя Франкленд и установил некоторые частные закономерности, его идеи не получили развития прежде всего потому, что он оставался приверженцем системы эквивалентов и пользовался неправильными значениями атомных масс. Решающую роль в создании теории валентности сыграл Фридрих Август Кекуле. В 1857 г. Кекуле в своей статье по-новому раскрыл теоретический смысл трёх основных типов Жерара – водорода, воды и аммиака: "Число атомов одного элемента, связанных с одним атомом другого, зависит от основности или величины сродства. Элементы в этом отношении распадаются на три главные группы: 1. Одноосновные или одноатомные (I), например H, Cl, Br, K. 2. Двухосновные или двухатомные (II), например O, S. 3. Трёхосновные или трёхатомные (III), например N, P, As. Отсюда вытекают три главных типа соединений: I + I, II + 2 I, III + 3 I, или же в виде простейших своих представителей HH, OH2, NH3". Здесь же Кекуле показал, что углерод является четырёхосновным (четырёхатомным) элементом, и его простейшим соединением является СН4 – отсюда и введённый им тип метана. В 1858 г. Кекуле развивает эти вопросы в статье "О конституции и превращениях химических соединений и о химической природе углерода". Уверенный в истинности своих представлений о валентности атомов, Кекуле ввёл их свой учебник органической химии, вышедший в 1859 г.: "Самый способ прилегания отдельных атомов внутри радикалов, так же как сложение атомов в соединения, обуславливается… основностью атомов". Основность, по мнению Кекуле – фундаментальное свойство атома, свойство такое же постоянное и неизменяемое, как и атомный вес. В 1858 г. взгляды, почти совпадающие с идеями Кекуле, высказал в статье "О новой химической теории" шотландец Арчибальд Скотт Купер. Однако, в отличие от Кекуле, Купер считал, что некоторые элементы способны проявлять переменную валентность: углерод характеризуется двумя степенями сродства – низшей в оксиде СО и высшей в диоксиде СО2. Купер предложил и выглядящие вполне современно (если отвлечься от удвоенного числа атомов кислорода вследствие признания за ним атомной массы, равной восьми) формулы некоторых органических соединений. Теории Кекуле и Купера с необыкновенной лёгкостью и простотой объясняли строение и сложных радикалов, и соединений в целом. Молекула любого химического соединения рассматривалась в этих теориях как целостное образование (дань унитарному учению Жерара), которое складывается из атомов за счёт полного взаимного насыщения единиц сродства. Теория валентности, несмотря на то, что Кекуле пришёл к ней, как к развитию теории типов Жерара-Лорана, совершенно иначе трактовала идею о целостности молекулы – лишь как неспособность её делиться на самостоятельно существующие радикалы. Иначе говоря, теория валентности поначалу представляла молекулу аддитивно – как сумму составляющих её атомов. Однако уже три года спустя, в сентябре 1861 г. русский химик Александр Михайлович Бутлеров внёс в теорию валентности важнейшие дополнения. В докладе "О химическом строении вещества", прочитанном на Съезде немецких естествоиспытателей и врачей в Шпейере, он изложил свою теорию химического строения. Основные положения этой теории Бутлеров сформулировал таким образом: 1) "Полагая, что каждому химическому атому свойственно лишь определённое и ограниченное количество химической силы (сродства), с которой он принимает участие в образовании тела, я назвал бы химическим строением эту химическую связь, или способ взаимного соединения атомов в сложном теле"; 2) "... Химическая натура сложной частицы определяется натурой элементарных составных частей, количеством их и химическим строением". Оставляя открытым вопрос о предпочтительном виде формул химического строения, Бутлеров так высказался об их смысле: "... когда сделаются известными общие законы зависимости химических свойств тел от их химического строения, то подобная формула будет выражением всех этих свойств". При этом Бутлеров был убеждён, что структурные формулы не могут быть просто условным изображением молекул, а должны отражать их реальное строение. Он подчёркивал, что каждая молекула имеет вполне определённую структуру и не может совмещать несколько таких структур. Бутлеров в своей теории проводил чёткое различие между свободным атомом и атомом, вступившим в соединение с другим, когда его сродство "связывается и переходит в новую форму". Бутлеров ввёл представление о полноте использования сил сродства и о "напряжении сродства", т. е. энергетической неэквивалентности связей, которая обусловлена взаимным влиянием атомов в молекуле. В результате этого взаимного влияния атомы в зависимости от их структурного окружения приобретают различное "химическое значение". Теория химического строения Бутлерова, в основе которой лежит теория валентности, позволила дать объяснение многим экспериментальным фактам, касавшимся изомерии органических соединений и их реакционной способности. Учебник Бутлерова "Введение к полному изучению органической химии" (1866) уже через год – впервые в истории отечественной науки – был опубликован на немецком языке. В 1862 г. немецкий химик Эмиль Эрленмейер пришёл к идее о существовании двойной связи углерод-углерод в этилене и тройной в ацетилене. Положение Бутлерова о взаимном влиянии атомов в молекуле активно развивал и другой русский химик – Владимир Васильевич Марковников – представивший в 1869 г. диссертацию "Материалы по вопросу о взаимном влиянии атомов в химических соединениях". Огромным достоинством теории химического строения явилась возможность наглядного изображения молекулы. История развития графического изображения валентности и химической связи сама по себе чрезвычайно интересна. Ниже приведены различные способы изображения формулы пропионовой кислоты в нескольких системах 1860-х годов.
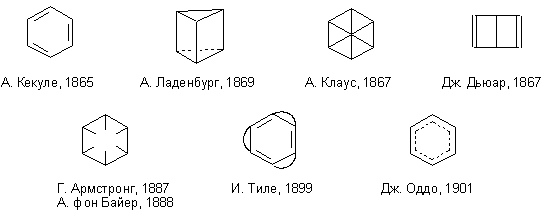
Т.н. "роликовые" модели, предложенные Кекуле в 1857 г., не были приняты научным сообществом, и сам автор от них быстро отказался. Некоторое время многими химиками использовались формулы, напоминающие формулы теории типов Жерара-Лорана, в которых связь между атомами и радикалами обозначалась с помощью фигурных скобок (одним из первых изображать в формулах теории типов чёрточку, соединяющую символы элементов, начал в 1864 г. Лотар Мейер). Шотландский химик Александер Крум Браун предложил в 1864 г. использовать структурные формулы в виде окружностей с помещёнными в них символами элементов, соединённых линиями, обозначающими химическую связь между атомами; количество линий соответствовало валентности атома. В 1865 г. немецкий химик-органик Август Вильгельм фон Гофман продемонстрировал первые шаростержневые модели, в которых роль атомов играли крокетные шары, получившие распространение под названием "глиптических" (глиптика – искусство резьбы по камню). В 1866 г. в учебнике Кекуле появились рисунки моделей, в которых атом углерода уже имел тетраэдрическую конфигурацию. Весьма важным для развития системы структурных формул стало установление строения молекулы бензола и, как следствие, огромного числа ароматических соединений. Замкнутое кольцо из шести атомов углерода, связанных попеременно одной или двумя валентностями предложил в 1865 г. Кекуле. Впрочем, попытки предложить формулу, объясняющую равноценность всех атомов углерода в молекуле бензола продолжались ещё долгое время.
Существенный вклад в развитие классической теории валентности в применении к ненасыщенным соединениям внёс немецкий химик Иоганнес Тиле, предложивший в 1899 г. теорию парциальных валентностей. Согласно этой теории, при образовании кратной связи между атомами углерода валентности этих атомов не насыщаются полностью, но каждый атом сохраняет свободной часть сродства – парциальную валентность. Если две двойные связи разделены одинарной, то образуется система сопряжённых двойных связей: парциальные валентности средних атомов углерода насыщаются, а парциальные валентности крайних остаются свободными. Теория парциальных валентностей удачно дополнила первоначальное учение о валентности, позволив дать объяснение многим реакциям, в частности, реакциям присоединения к ненасыщенным углеводородам. Ещё одной попыткой усовершенствования теории валентности стала теория мезогидрии, которую разработал итальянский химик Джузеппе Оддо. Оддо предположил, что атом водорода, находясь вблизи двух атомов многовалентных элементов, может делить свою валентность между ними; его теория позволила дать объяснение, в частности, явлению таутомерии. Важнейшим этапом развития структурной химии стало установления пространственного строения молекул. Ещё в 1867 г. Кекуле критиковал формулы Крум-Брауна, поскольку считал, что такой вид изображения даёт неверное представление, будто все валентности находятся в одной плоскости. Сам Кекуле неоднократно высказывал предположение о тетраэдрическом расположении четырёх валентностей углерода. Возникновение стереохимии было обусловлено целым рядом открытий, поначалу, казалось, не имевших отношения к химии. В 1801 г. Томас Юнг провёл опыты, доказавшие волновую природу света. Огюстен Жан Френель около 1814 г. показал, что световые волны относятся к типу поперечных волн, объяснив природу плоскополяризованного света, который открыл в 1808 г. Этьен Луи Малюс. В 1815 г. французский физик Жан Батист Био открыл явление оптической активности некоторых веществ. Выделенные Берцелиусом в 1832 г. изомерные винная и виноградная кислоты (винная вращает плоскость поляризации света вправо, виноградная оптически неактивна) стали первым примером оптической изомерии.
В 1848 г. французский химик и биолог Луи Пастер обнаружил, что виноградная кислота представляет собой смесь двух оптических антиподов – винной кислоты и её изомера, отличающегося лишь тем, что он вращает плоскость поляризации света влево. Кристаллы этих изомеров оказались асимметричными; Пастер высказал предположение о том, что асимметрия кристаллов и оптическая активность должны быть обусловлена асимметрией молекул. Иоганн Адольф Вислиценус, открывший изомеры молочной кислоты, опять-таки различающиеся только оптической активностью, писал в 1873 г.: "различие оптических изомеров можно объяснить различным расположением их атомов в пространстве, и надлежит искать определённых представлений об этом расположении".
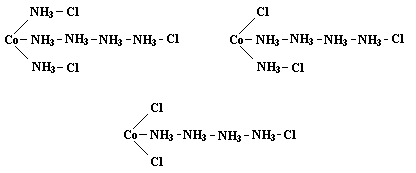
Гипотезу, объясняющую оптическую изомерию – гипотезу асимметричного атома углерода – предложил в 1874 г. датский химик Якоб Генрик Вант-Гофф. Вант-Гофф основывался на предположении о том, что четыре валентности атома углерода направлены из центра тетраэдра к вершинам. Симметрия нарушается в том случае, когда все четыре атома или радикала, соединённые с данным атомом углерода, различны. Присоединение в этом случае может произойти двумя различными способами, и полученные фигуры представляют собой зеркальное отображение друг друга – как раз тот тип асимметрии, который наблюдал Пастер в кристаллах винных кислот. Почти одновременно с Вант-Гоффом подобные предположения опубликовал французский химик Жозеф Ашиль Ле Бель. Гипотеза Вант-Гоффа – Ле Беля очень удачно предсказывала оптическую активность соединений и количество оптических изомеров. В 1875 г. в новом издании своей брошюры "Химия в пространстве" Вант-Гофф дал объяснение оптической активности соединений алленового типа, показав, что эти вещества обладают асимметричным строением и должны поэтому иметь правую и левую формы даже при отсутствии асимметричного атома углерода. Несмотря на блестящее совпадение выводов Вант-Гоффа с опытными данными, новая теория (активно пропагандируемая Вислиценусом) вызвала ожесточённую дискуссию; Кекуле отнёсся к ней весьма скептически, а выдающийся немецкий химик-органик Адольф Вильгельм Герман Кольбе выступил в 1877 г. с чрезвычайно резкой критикой в адрес Вант-Гоффа и Вислиценуса. Тем не менее, даже авторитет Кольбе оказался не в силах затмить несомненные достоинства теории. В защиту стереохимии выступили Вильгельм Фридрих Оствальд, Шарль Адольф Вюрц, Альберт Ладенбург. В 1885 г. немецкий химик Иоганн Фридрих Вильгельм Адольф фон Байер суммировал положения классической теории валентности и стереохимии, разработав теорию напряжения, призванной объяснить устойчивость циклических и непредельных соединений, в основе которой лежит представление о тетраэдрическом углеродном атоме. Вислиценус в 1887 г. на основе теории Вант-Гоффа дал надлежащее объяснение геометрической изомерии на примере малеиновой и фумаровой кислот. Окончательное утверждение теории Вант-Гоффа состоялось в 1890-х гг. благодаря блестящим синтетическим работам, которые выполнили Пауль Вальден, открывший т.н. вальденовское обращение, и Эмиль Герман Фишер. На протяжении довольно долгого времени теория валентности применялась главным образом к органическим соединениям. Однако довольно скоро структурные представления оказались востребованы также и в химии комплексных соединений. Теоретические представления этого раздела неорганической химии формировались на основе изучения свойств комплексов, получаемых взаимодействием солей переходных металлов с аммиаком. Первым шагом на пути к координационной химии стала аммонийная гипотеза Томаса Грэма (1840 г.), усмотревшего аналогию между взаимодействием аммиака с кислотами и с солями металлов; согласно этой гипотезе, металл занимал место одного из атомов водорода в ионе аммония. Гипотеза Грэма была развита в 1851 г. Гофманом, предположившим, что атом водорода в аммонийном радикале способен замещаться на другой аммонийный радикал. Следующим шагом стала цепная теория, предложенная в 1869 г. Кристианом Вильгельмом Бломстрандом и усовершенствованная Софусом Мадсом Йёргенсеном. В теории Бломстранда – Йёргенсена для некоторых элементов допускалась валентность выше обычной, а также возможность образования цепей атомами азота, кислорода и других элементов. Экспериментально установленное различие между кислотными остатками, входящими в состав комплекса, объяснялось различным способом их связывания – непосредственно с металлом или с концом цепи. Например, для аммиакатных комплексов состава CoCl3·6NH3, CoCl3·5NH3 и CoCl3·4NH3, из растворов которых нитратом серебра осаждаются три, два и один эквивалент хлора соответственно, Йёргенсен предполагал следующее строение:
Однако теория Бломстранда – Йёргенсена не могла объяснить, например, существование двух изомерных комплексов состава CoCl3·4NH3 – празеосоли (зелёный) и виолеосоли (фиолетовый). В 1893 г. швейцарский химик Альфред Вернер опубликовал статью "О строении неорганических соединений", в которой изложил основные положения созданной им координационной теории. Теория Вернера (который уже был известен своими работами по стереохимии органических соединений азота) существенно расширила представления химиков о валентности элементов. Главное положение теории состояло в признании за атомами способности, наряду со связями сродства (главная, первичная или обычная валентность), образовывать дополнительные (координационные) связи (побочная или вторичная валентность). Вернер ввёл понятие о комплексном ионе – сложной комбинации атомов и молекул, состоящем из центрального иона, связанного с несколькими молекулами или отрицательными ионами (лигандами), образующими внутреннюю сферу комплекса; главной характеристикой центрального иона Вернер считал координационное число, равное сумме главной и побочной валентностей. Для упоминавшихся выше комплексных солей кобальта в рамках координационной теории предполагалось строение с координационным числом 6: [Co(NH3)6]Cl3 [Co(NH3)5Cl]Cl2 [Co(NH3)4Cl2]Cl
Важной частью координационной теории стали положения, касающиеся стереохимии комплексных соединений. Вернер предположил, что для комплексов с координационным числом 6 валентности направлены к вершинам октаэдра, в центре которого находится центральный ион; для комплексов с координационным числом 4 валентности направлены либо к вершинам тетраэдра, либо к углам квадрата. В связи с этими положениями Вернер подробно рассмотрел возможные случаи изомерии соответствующих комплексов, в частности, цис-транс-изомерии в квадратных и октаэдрических комплексах и оптической изомерии в тетраэдрических и октаэдрических комплексах. Теория Вернера наглядно объяснила строение комплексных соединений, легла в основу их классификации и позволила предсказать существование большого числа новых соединений. Слабой стороной теории являлись положения о двух типах валентности, которые невозможно было обосновать теоретически. Это сделало ещё более острой главную проблему структурной химии XIX века (да и всей атомно-молекулярной теории): после отказа от электрохимического дуализма Берцелиуса в распоряжении учёных не имелось хоть сколько-нибудь адекватных представлений о природе валентных сил. Блестящие успехи химической теории, казалось, нисколько не приблизили естествоиспытателей к ответу на вопрос о способе соединения атомов. Как и в случае периодического закона, ответ оказался за пределами предмета химии. Предыдущая глава В начало страницы Следующая глава
|